spectralClones - Spectral clustering method for clonal partitioning
Description¶
spectralClones provides an unsupervised spectral clustering
approach to infer clonal relationships in high-throughput Adaptive Immune Receptor
Repertoire sequencing (AIRR-seq) data. This approach clusters B or T cell receptor
sequences based on junction region sequence similarity and shared mutations within
partitions that share the same V gene, J gene, and junction length, allowing for
ambiguous V or J gene annotations.
Usage¶
spectralClones(
db,
method = c("novj", "vj"),
germline = "germline_alignment",
sequence = "sequence_alignment",
junction = "junction",
v_call = "v_call",
j_call = "j_call",
clone = "clone_id",
fields = NULL,
cell_id = NULL,
locus = "locus",
only_heavy = TRUE,
split_light = TRUE,
targeting_model = NULL,
len_limit = NULL,
first = FALSE,
cdr3 = FALSE,
mod3 = FALSE,
max_n = 0,
threshold = NULL,
base_sim = 0.95,
iter_max = 1000,
nstart = 1000,
nproc = 1,
verbose = FALSE,
log = NULL,
summarize_clones = TRUE
)
Arguments¶
- db
- data.frame containing sequence data.
- method
- one of the
"novj"or"vj". See Details for description. - germline
- character name of the column containing the germline or reference sequence.
- sequence
- character name of the column containing input sequences.
- junction
- character name of the column containing junction sequences. Also used to determine sequence length for grouping.
- v_call
- name of the column containing the V-segment allele calls.
- j_call
- name of the column containing the J-segment allele calls.
- clone
- output column name containing the clonal cluster identifiers.
- fields
- character vector of additional columns to use for grouping. Sequences with disjoint values in the specified fields will be classified as separate clones.
- cell_id
- name of the column containing cell identifiers or barcodes.
If specified, grouping will be performed in single-cell mode
with the behavior governed by the
locusandonly_heavyarguments. If set toNULLthen the bulk sequencing data is assumed. - locus
- name of the column containing locus information.
Only applicable to single-cell data.
Ignored if
cell_id=NULL. - only_heavy
- use only the IGH (BCR) or TRB/TRD (TCR) sequences
for grouping. Only applicable to single-cell data.
Ignored if
cell_id=NULL. - split_light
- split clones by light chains. Ignored if
cell_id=NULL. - targeting_model
- TargetingModel object. Only applicable if
method="vj". See Details for description. - len_limit
- IMGT_V object defining the regions and boundaries of the Ig
sequences. If NULL, mutations are counted for entire sequence. Only
applicable if
method="vj". - first
- specifies how to handle multiple V(D)J assignments for initial grouping.
If
TRUEonly the first call of the gene assignments is used. IfFALSEthe union of ambiguous gene assignments is used to group all sequences with any overlapping gene calls. - cdr3
- if
TRUEremoves 3 nucleotides from both ends of"junction"prior to clustering (converts IMGT junction to CDR3 region). IfTRUEthis will also remove records with a junction length less than 7 nucleotides. - mod3
- if
TRUEremoves records with ajunctionlength that is not divisible by 3 in nucleotide space. - max_n
- the maximum number of degenerate characters to permit in the junction sequence before excluding the
record from clonal assignment. Default is set to be zero. Set it as
"NULL"for no action. - threshold
- the supervising cut-off to enforce an upper-limit distance for clonal grouping. A numeric value between (0,1).
- base_sim
- required similarity cut-off for sequences in equal distances from each other.
- iter_max
- the maximum number of iterations allowed for kmean clustering step.
- nstart
- the number of random sets chosen for kmean clustering initialization.
- nproc
- number of cores to distribute the function over.
- verbose
- if
TRUEprints out a summary of each step cloning process. ifFALSE(default) process cloning silently. - log
- output path and filename to save the
verboselog. The input file directory is used if path is not specified. The default isNULLfor no action. - summarize_clones
- if
TRUEperforms a series of analysis to assess the clonal landscape and returns a ScoperClones object. IfFALSEthen a modified inputdbis returned. When grouping byfields,summarize_clonesshould beFALSE.
Value¶
If summarize_clones=TRUE (default) a ScoperClones object is returned that includes the
clonal assignment summary information and a modified input db in the db slot that
contains clonal identifiers in the specified clone column.
If summarize_clones=FALSE modified data.frame is returned with clone identifiers in the
specified clone column.
Details¶
If method="novj", then clonal relationships are inferred using an adaptive
threshold that indicates the level of similarity among junction sequences in a local neighborhood.
If method="vj", then clonal relationships are inferred not only on
junction region homology, but also taking into account the mutation profiles in the V
and J segments. Mutation counts are determined by comparing the input sequences (in the
column specified by sequence) to the effective germline sequence (IUPAC representation
of sequences in the column specified by germline).
While not mandatory, the influence of SHM hot-/cold-spot biases in the clonal inference
process will be noted if a SHM targeting model is provided through the targeting_model
argument. See TargetingModel for more technical details.
If the threshold argument is specified, then an upper limit for clonal grouping will
be imposed to prevent sequences with dissimilarity above the threshold from grouping together.
Any sequence with a distance greater than the threshold value from the other sequences,
will be assigned to a singleton group.
Single-cell data¶
To invoke single-cell mode the cell_id argument must be specified and the locus
column must be correct. Otherwise, clustering will be performed with bulk sequencing assumptions,
using all input sequences regardless of the values in the locus column.
Values in the locus column must be one of c("IGH", "IGI", "IGK", "IGL") for BCR
or c("TRA", "TRB", "TRD", "TRG") for TCR sequences. Otherwise, the operation will exit and
return an error message.
Under single-cell mode with paired-chain sequences, there is a choice of whether
grouping should be done by (a) using IGH (BCR) or TRB/TRD (TCR) sequences only or
(b) using IGH plus IGK/IGL (BCR) or TRB/TRD plus TRA/TRG (TCR) sequences.
This is governed by the only_heavy argument. There is also choice as to whether
inferred clones should be split by the light/short chain (IGK, IGL, TRA, TRG) following
heavy/long chain clustering, which is governed by the split_light argument.
In single-cell mode, clonal clustering will not be performed on data were cells are
assigned multiple heavy/long chain sequences (IGH, TRB, TRD). If observed, the operation
will exit and return an error message. Cells that lack a heavy/long chain sequence (i.e., cells with
light/short chains only) will be assigned a clone_id of NA.
Examples¶
# Subset example data
db <- subset(ExampleDb, c_call == "IGHG")
# Find clonal groups
results <- spectralClones(db, method="novj", germline="germline_alignment_d_mask")
Running defineClonesScoper in bulk mode and only keep heavy chains
# Retrieve modified input data with clonal clustering identifiers
df <- as.data.frame(results)
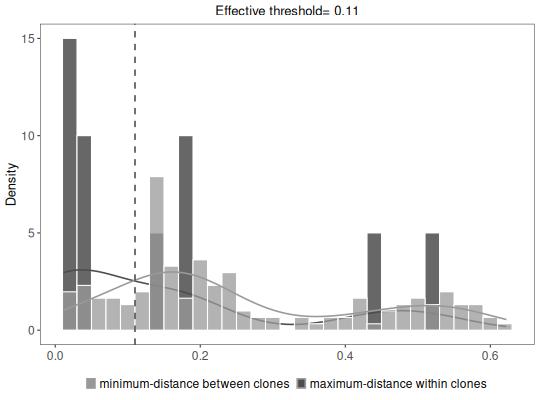
# Plot clonal summaries
plot(results, binwidth=0.02)
See also¶
See plotCloneSummary for plotting summary results. See groupGenes for more details about grouping requirements.